Introduction
Although drugs, alternative splicing, and neoantigens are intimately interconnected, existing resources fail to integrate these three dimensions. To bridge this gap, we constructed DRIVE (Drug-induced RNA profile, Isoform Variability and Epitopes), a comprehensive resource integrating, drug induced transcriptomic responses, alternative splicing and neoantigen landscapes. We systematically retrieved drug-treated and matched control cancer cell line transcriptomic datasets from the GEO database, utilizing Large Language Models (LLMs) combined with rigorous manual inspection to ensure high-fidelity metadata. By implementing a standardized pipeline for raw data processing, we quantified drug-induced differential gene expression, AS alterations, and predicted splice-neoantigens across thousands of conditions.
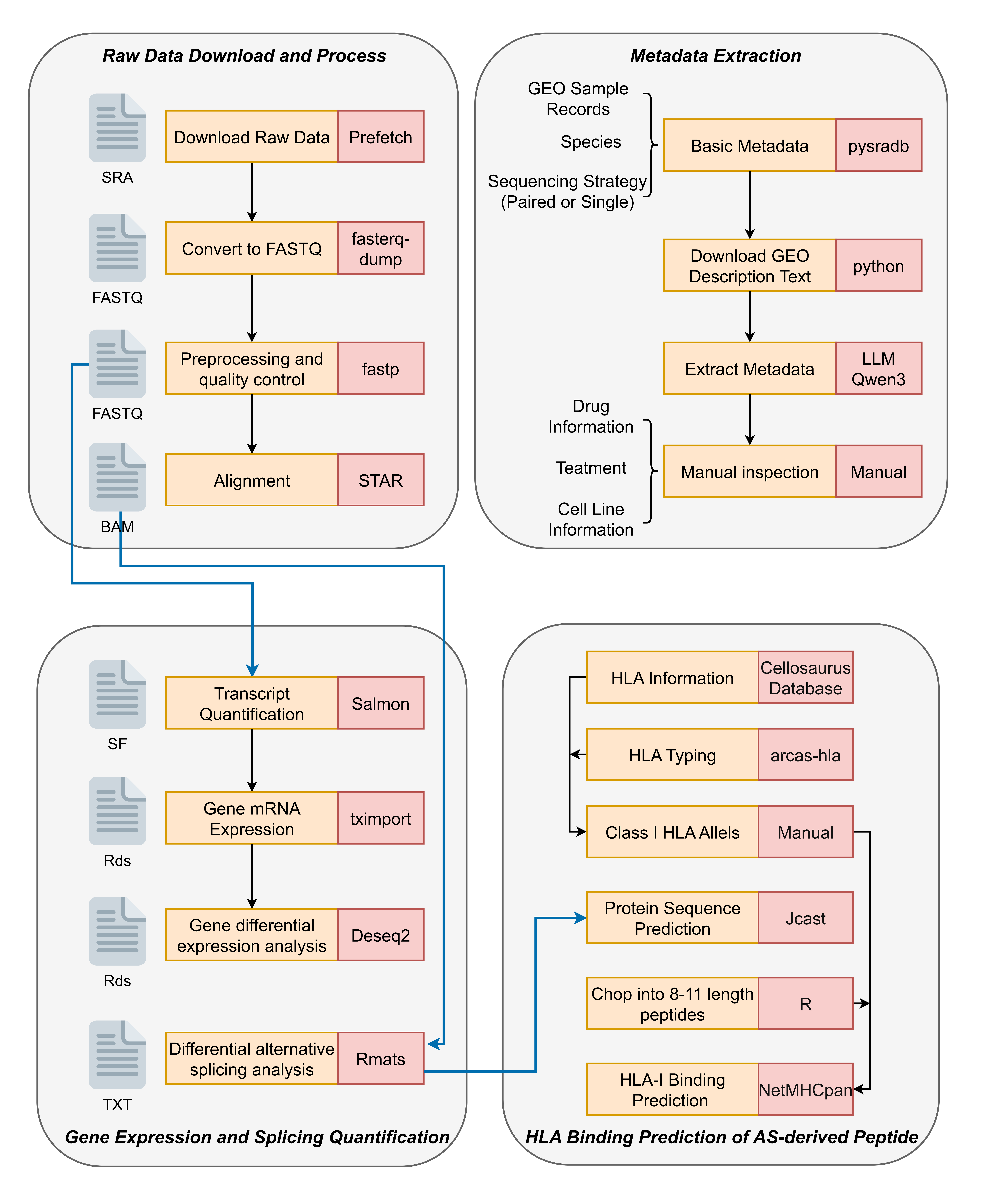
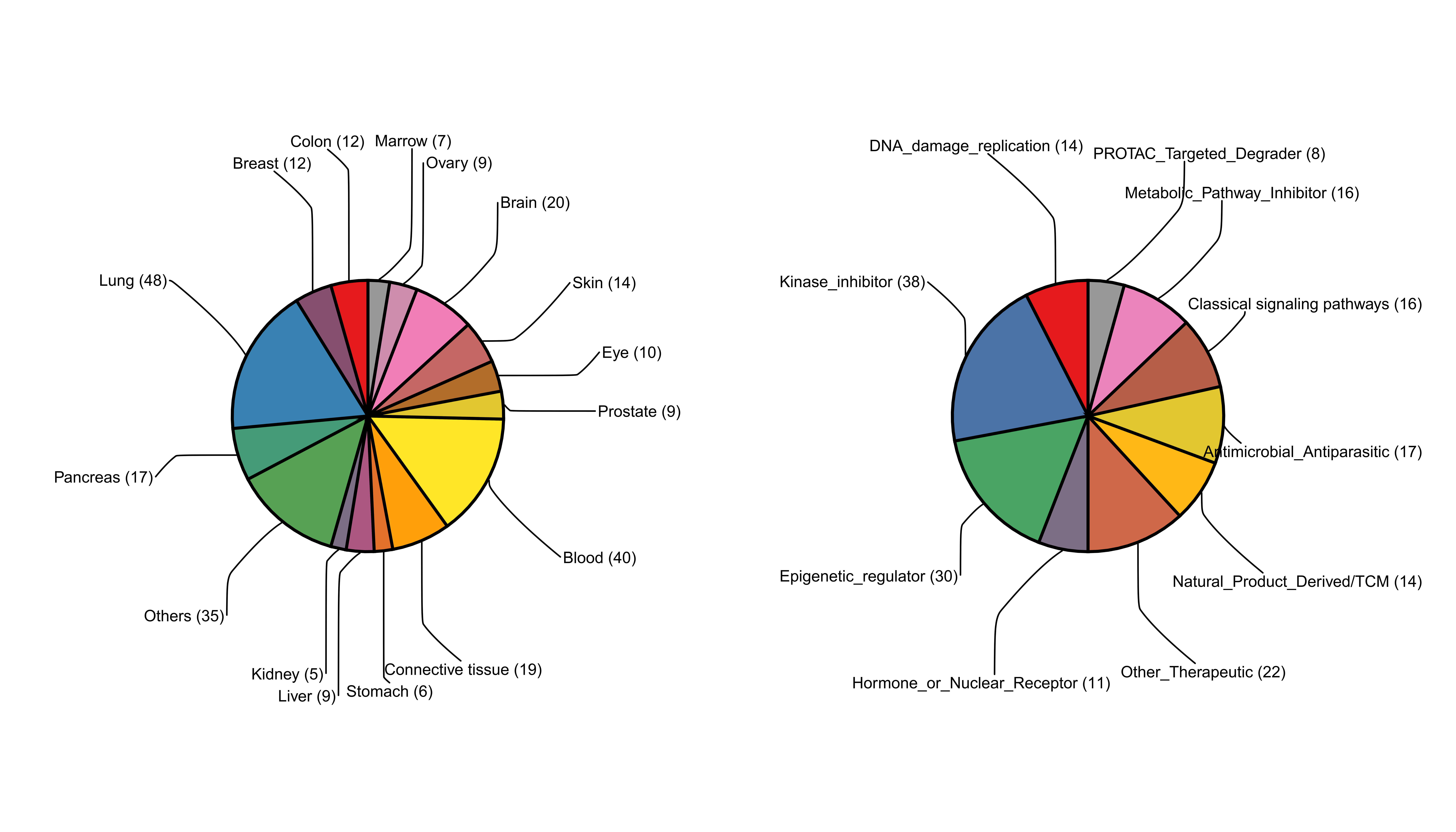
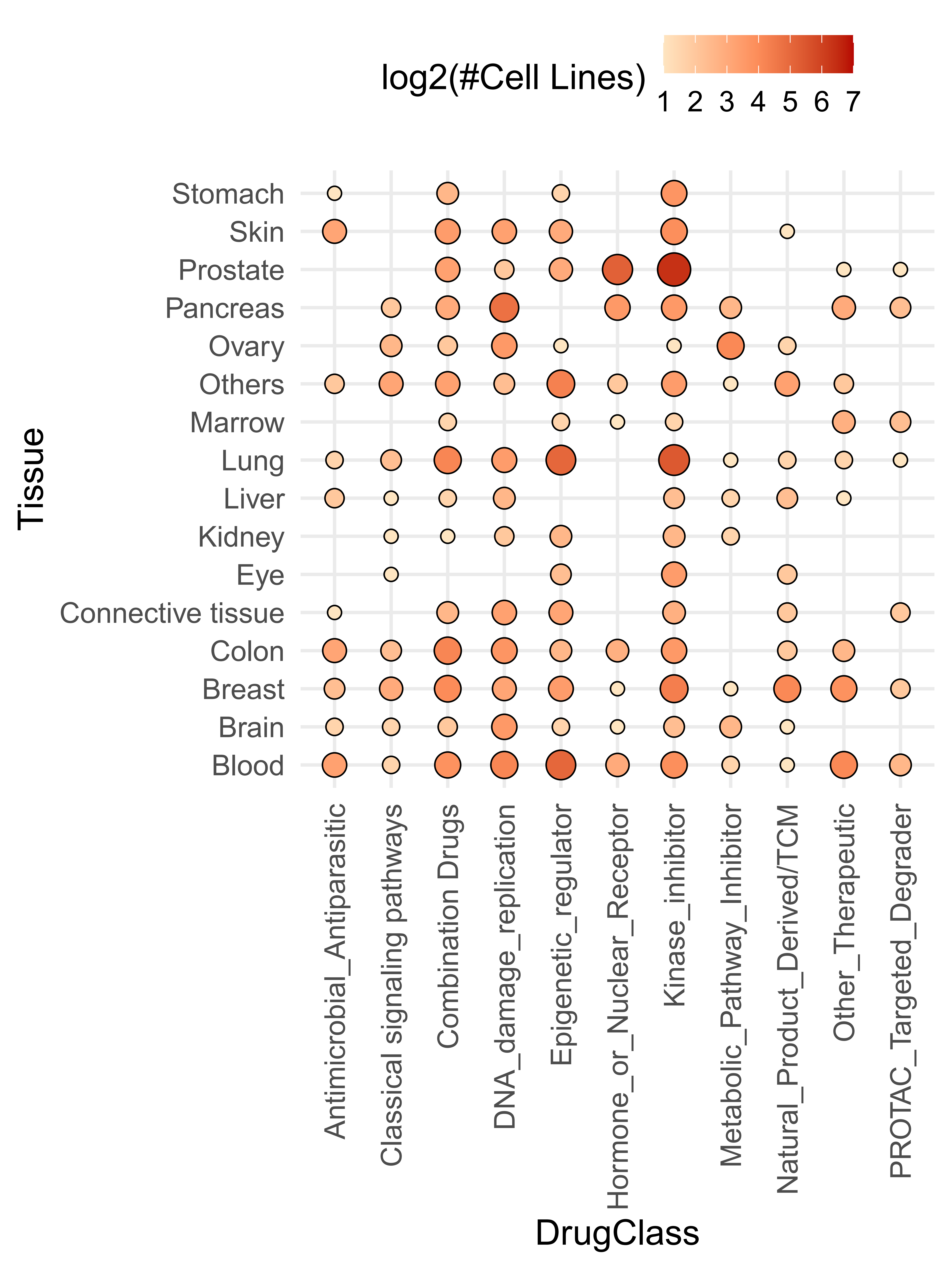
Following the careful manual inspection, the retained high-confidence datasets were subjected to further analysis .The final data consist of 3,911 samples from 243 datasets, involving 931 drug-cell treatments from 272 cancer cell lines and 278 treated drugs.
Transcriptome response
In the Transcriptome Response module, users can select drug names and cell line names to display information about the corresponding drug-treated samples. This module presents information in three areas: Metadata, Gene Expression, and Alternative Splicing.
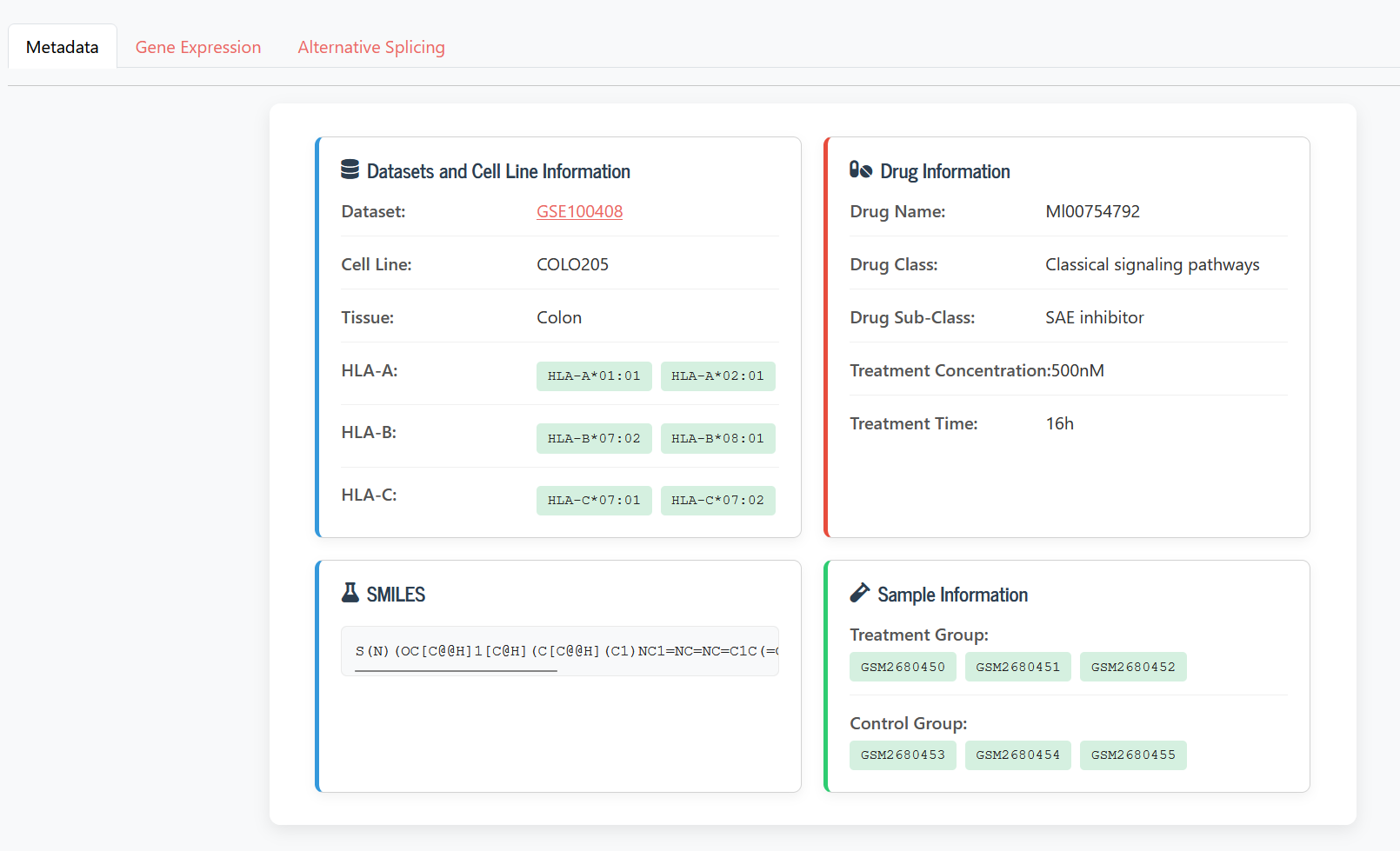
Metadata displays specific details about the cell lines and drug treatments.
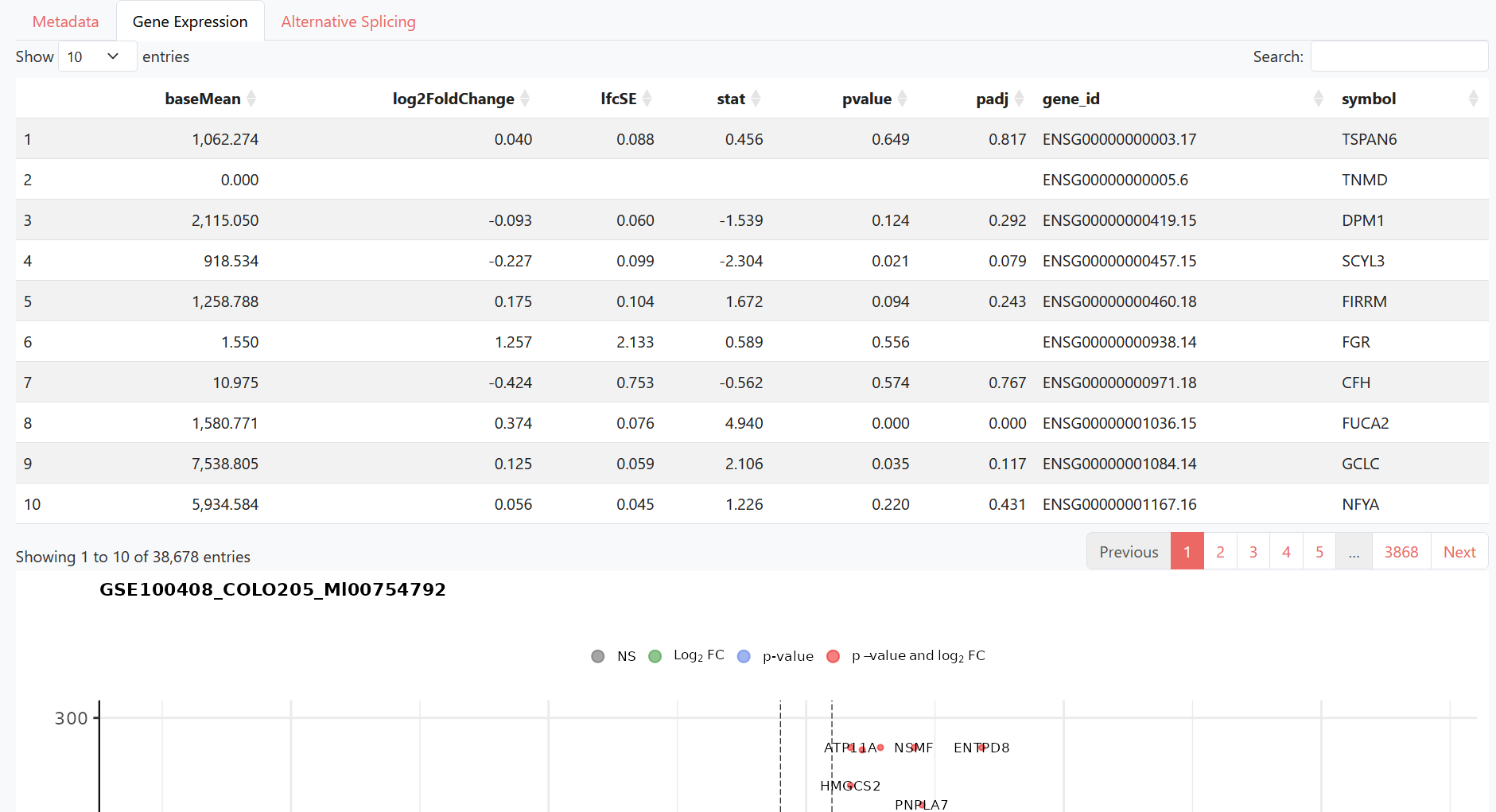
Gene Expression shows the results of differential expression analysis (including gene lists and volcano plots).
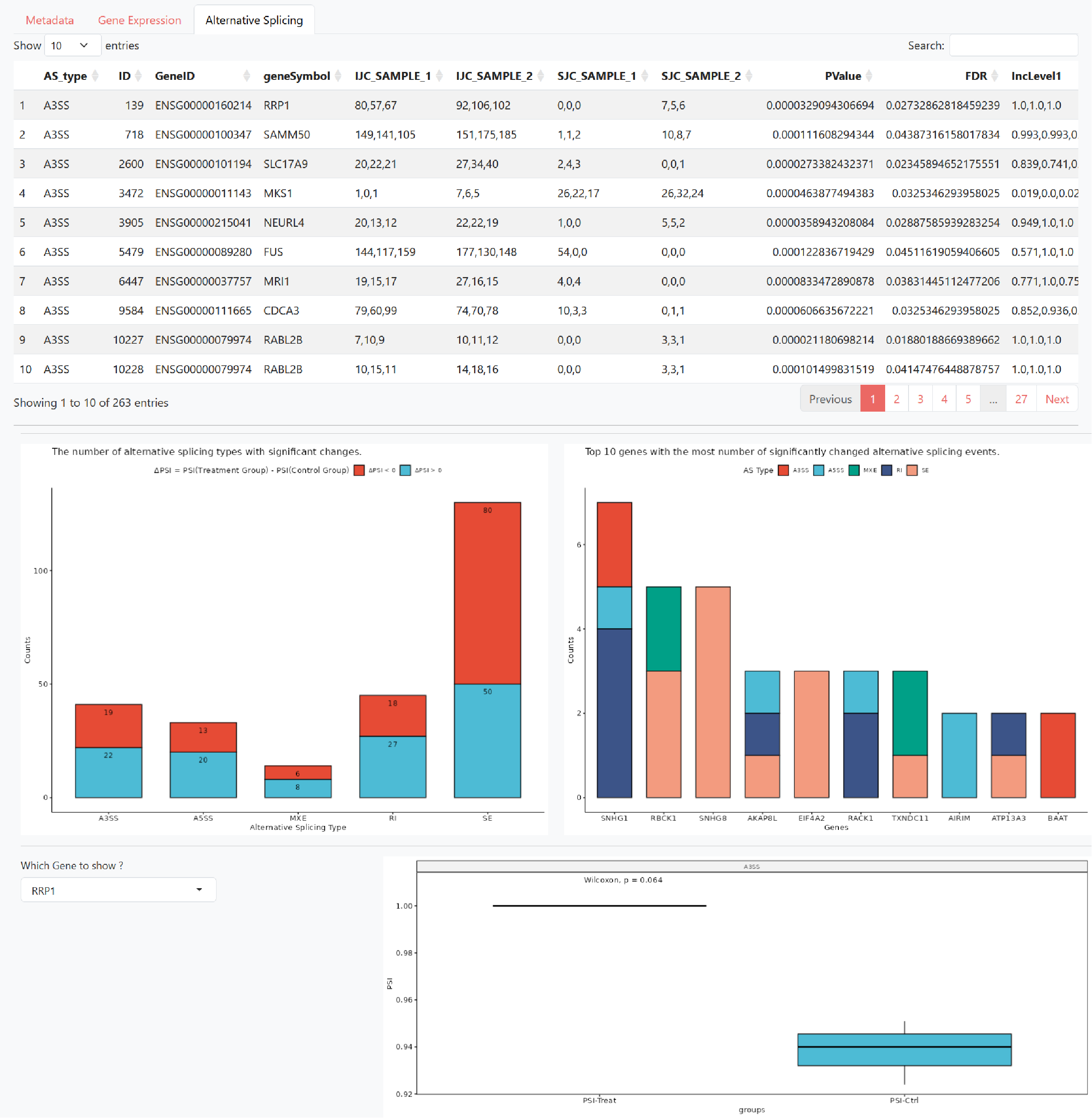
Alternative Splicing presents the results of differential alternative splicing analysis (rMATS), which includes:
A table of differential alternative splicing events (FDR < 0.05)
Statistics on the number of significantly different alternative splicing types (PSI corresponds to the IncLevelDifference column in rMATS output, PSI of the treatment group corresponds to IncLevel1, and PSI of the control group corresponds to IncLevel2)
The top 10 genes with the highest number of significant alternative splicing events
PSI differences between the Treatment and Control groups based on the user-selected gene
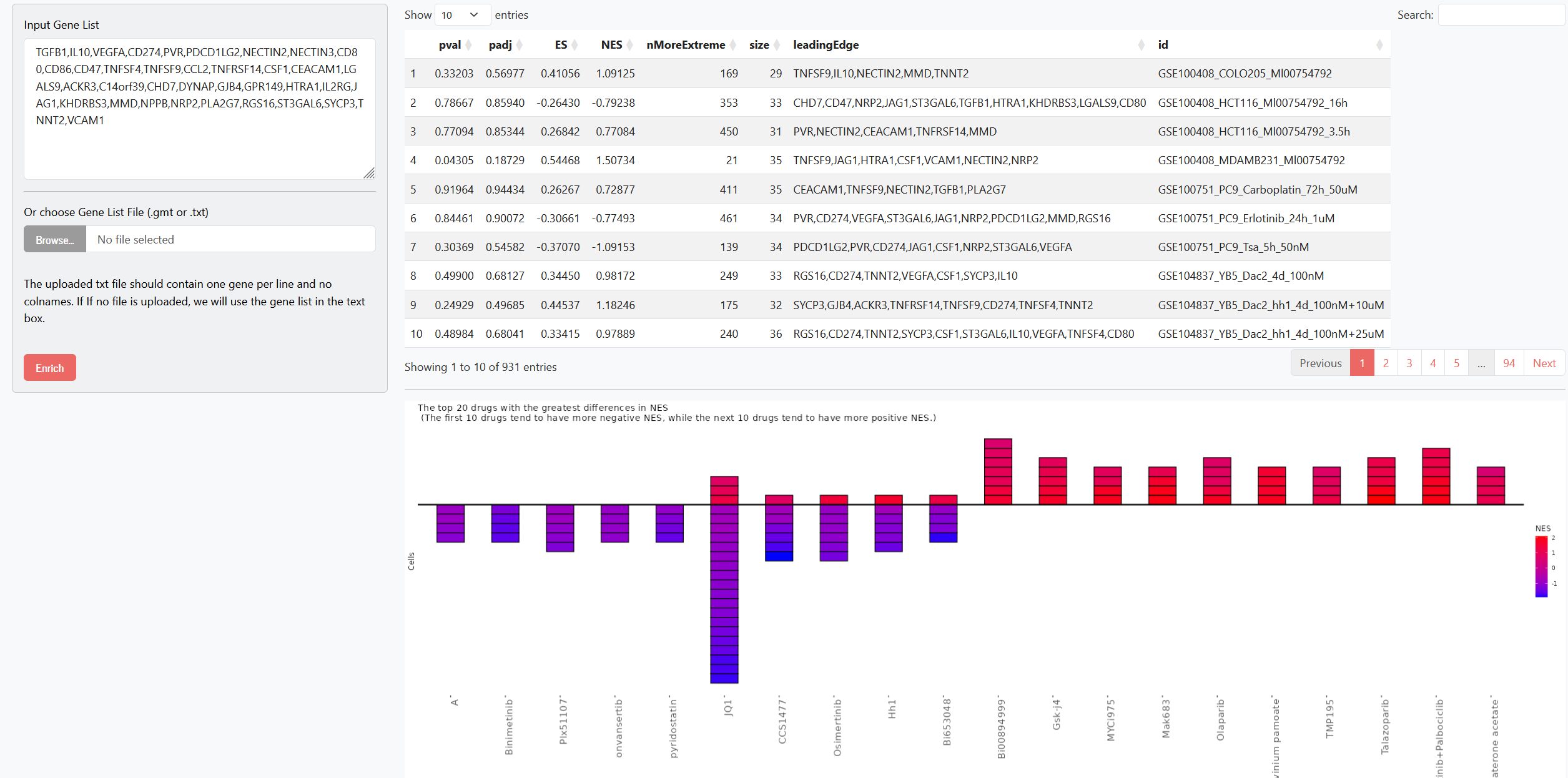
Drug Enrichment
In the Drug Enrichment module, users can upload a gene list (via direct text input or by uploading gmt/txt files). We perform GSEA analysis based on the user-uploaded gene set to identify drug–cell line samples that are significantly enriched for that gene set. The module displays the enrichment results table, along with the top 10 drugs showing positively biased enrichment and the top 10 showing negatively biased enrichment. In the visualization, each small square represents a cell line sample, and the color intensity corresponds to the magnitude of the NES (Normalized Enrichment Score).
Connectivity Analysis
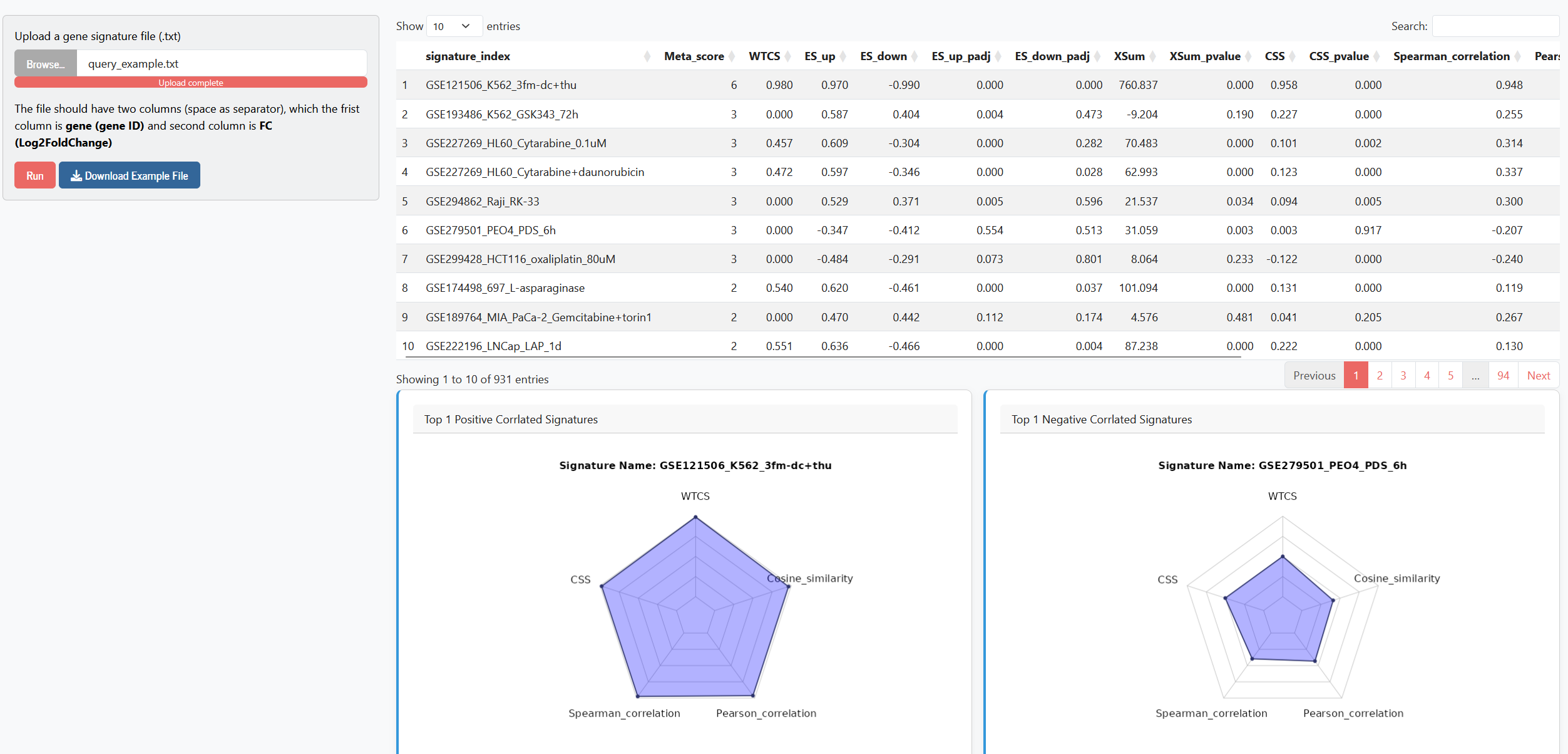
In the Connectivity Analysis module, users can provide a gene signature file. The file should have two columns (space as separator), with the first column being the gene (gene ID) and the second column being FC (Log2FoldChange). We screen the database for drug-induced gene expression signatures that have high similarity with the user-provided query signature. Adopting the strategy and source code from the WebCMap R package, we computed six similarity metrics: eXtreme Sum (XSum), Connection Strength Score (CSS), Weighted Connectivity Score (WTCS), Spearman correlation, Pearson correlation, and Cosine similarity. For CSS and XSum, empirical P-values are determined using a permutation test. Similar to the approach used in WebCMap, we pre-generated 10,000 random sets for each of the 931 signatures in our DRIVE dataset and calculated their corresponding CSS and XSum values in advance, thereby constructing the null distributions required for the empirical P-value calculations. A table showing all similarity metrics between the drug-induced signatures and the user-provided signature is displayed, along with radar charts for the top-ranked (positively and negatively correlated) drug-induced signatures (note that XSum values are not shown in the radar charts due to inconsistent value ranges).
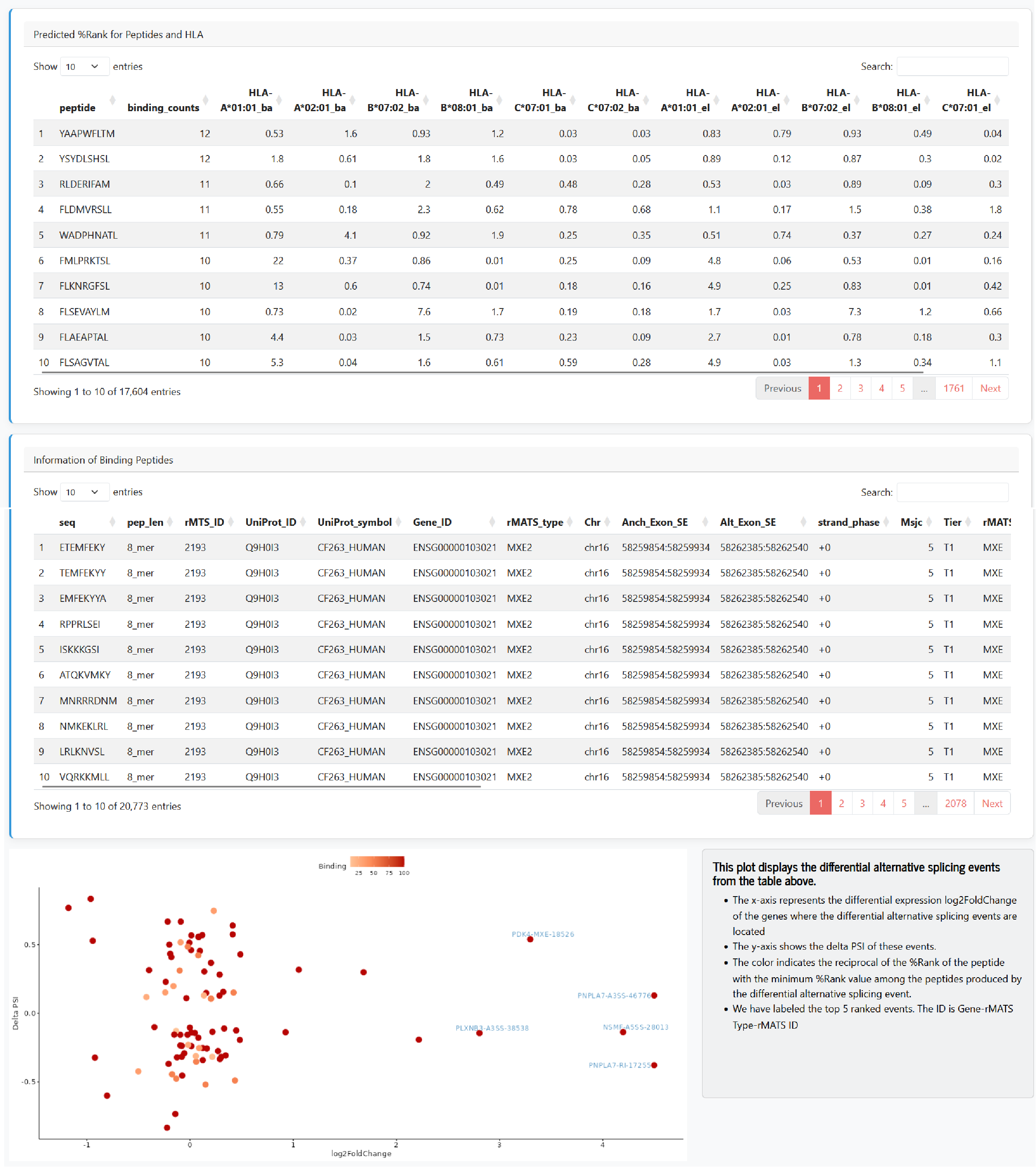
AS-derived Neoantigen
In this module, users can select a drug name and a cell line name to display neoantigen information derived from significantly differential alternative splicing events (FDR < 0.05 and absolute delta PSI > 0.1) in the corresponding drug-treated samples. We define peptides that bind to HLA as potential binders if both the predicted %Rank_BA and %Rank_EL scores for at least one HLA allele are less than 0.5 (or %Rank < 2 for exploratory analysis in the Shiny web server). A table is displayed showing the predicted %Rank scores for binding peptides and each HLA allele, as well as a table of information for each binding peptide (including the source rMATS ID, rMATS Type, gene, etc.). We also present a ternary relationship diagram illustrating the connections between alternative splicing, differential expression, and binding peptides.